Introduction
The Jupyter Notebook is an open-source web application that allows you to create and share documents that contain code, equations, visualizations and text. The functionality is partly overlapping with R Markdown (see the tutorial), in that they both use markdown and code chunks to generate reports that integrate results of computations with the code that generated them. Jupyter Notebook comes from the Python community while R Markdown was developed by RStudio, but you could use most common programming languages in either alternative. In practice though, it's quite common that R developers use Jupyter but probably not very common that Python developers use RStudio. Some reasons to use Jupyter include:
- Python is lacking a really good IDE for doing exploratory scientific data analysis, like RStudio or Matlab. Some people use Jupyter simply as an alternative for that.
- The community around Jupyter notebooks is large and dynamic, and there are lots of tools for sharing, displaying or interacting with notebooks.
- An early ambition with Jupyter notebooks (and its predecessor IPython notebooks) was to be analogous to the lab notebook used in a wet lab. It would allow the data scientist to document his or her day-to-day work and interweave results, ideas, and hypotheses with the code. From a reproducibility perspective, this is one of the main advantages.
- Jupyter notebooks can be used, just like R Markdown, to provide a tighter connection between your data and your results by integrating results of computations with the code that generated them. They can also do this in an interactive way that makes them very appealing for sharing with others.
As always, the best way is to try it out yourself and decide what to use it for! Here are some useful resources if you want to read more:
- The Jupyter project site contains a lot of information and inspiration.
- The Jupyter Notebook documentation.
- A guide to using widgets for creating interactive notebooks.
This tutorial depends on files from the course GitHub repo. Take a look at the
setup for instructions on how to set it up if you haven't done so
already. Then open up a terminal and go to workshop-reproducible-research/jupyter
and activate your jupyter-env Conda environment.
A note on nomenclature
- Jupyter: a project to develop open-source software, open-standards, and services for interactive computing across dozens of programming languages. Lives at jupyter.org.
- Jupyter Notebook: A web application that you use for creating and managing notebooks. One of the outputs of the Jupyter project.
- Jupyter notebook: The actual
.ipynbfile that constitutes your notebook.
Getting started
One thing that sets Jupyter Notebook apart from what you might be used to is that it's a web application, i.e. you edit and run your code from your browser. But first you have to start the Jupyter Notebook server.
$ jupyter notebook --allow-root
[I 18:02:26.722 NotebookApp] Serving notebooks from local directory: /Users/john/Documents/projects/workshop-reproducible-research/jupyter
[I 18:02:26.723 NotebookApp] 0 active kernels
[I 18:02:26.723 NotebookApp] The Jupyter Notebook is running at:
[I 18:02:26.723 NotebookApp] http://localhost:8888/?token=e03f10ccb40efc3c6154358593c410a139b76acf2cae785c
[I 18:02:26.723 NotebookApp] Use Control-C to stop this server and shut down all kernels (twice to skip confirmation).
[C 18:02:26.724 NotebookApp]
Copy/paste this URL into your browser when you connect for the first time,
to login with a token:
http://localhost:8888/?token=e03f10ccb40efc3c6154358593c410a139b76acf2cae785c
[I 18:02:27.209 NotebookApp] Accepting one-time-token-authenticated connection from ::1
Jupyter Notebook probably opened up a web browser for you automatically,
otherwise go to the address specified in the message in the terminal. Note that
the server is running locally (as http://localhost:8888) so this does not
require that you have an active internet connection. Also note that it says:
Serving notebooks from local directory: </some/local/path/workshop-reproducible-research/jupyter>
Everything you do in your Notebook session will be stored in this directory, so you won't lose any work if you shut down the server.
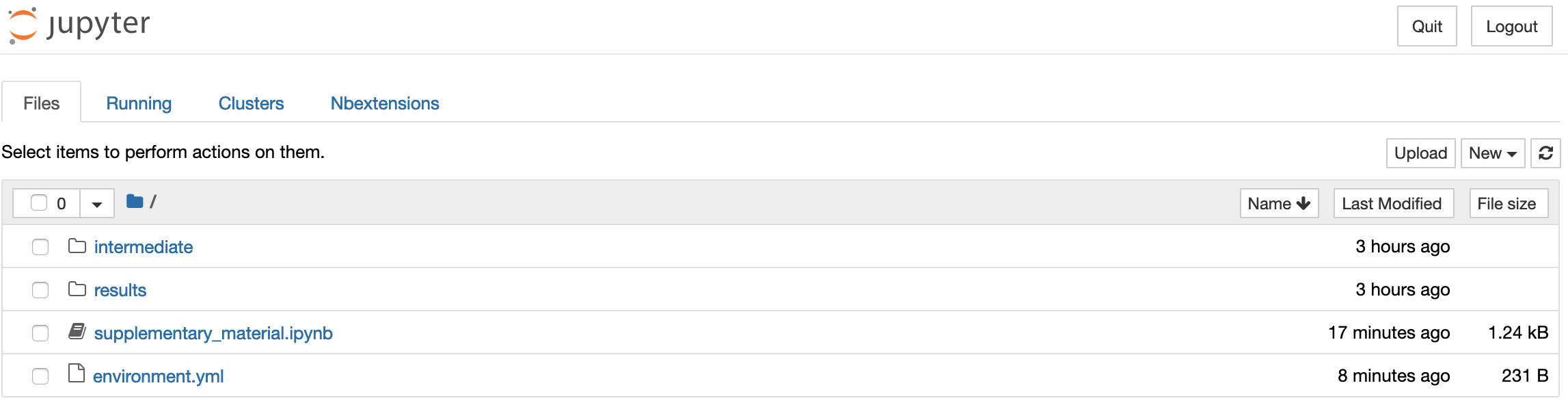
What you're looking at is the Notebook dashboard. This is where you manage your files, notebooks, and kernels. The Files tab shows the files in your directory. The Running tab keeps track of all your processes. The third tab, Clusters, is used for parallel computing and won't be discussed further in this tutorial. Finally, the Nbextensions tab shows a list of configurable notebook extensions that you can use to add functionality to your notebook (as we'll see below).
Let's start by creating an empty notebook by selecting the Files tab and clicking New > Notebook > Python 3. This will open up a new tab or window looking like this:
![]()
Tip
If you want to start Jupyter Notebooks on a cluster that you SSH to (e.g. Uppmax) see the section in the Extra material
The basics
Jupyter notebooks are made up of cells, and you are currently standing in the first cell in your notebook. The fact that it has a green border indicates that it's in "Edit mode", so you can write stuff in it. A blue border indicates "Command mode" (see below). Cells in Jupyter notebooks can be of two types: markdown or code.
- Markdown: These cells contain static material such as captions, text, lists, images and so on. You express this using Markdown, which is a lightweight markup language. Markdown documents can then be converted to other formats for viewing (the document you're reading now is written in Markdown and then converted to HTML). The format is discussed a little more in detail in the R Markdown tutorial. Jupyter Notebook uses a dialect of Markdown called Github Flavored Markdown, which is described here.
- Code: These are the cells that actually do something, just as code chunks do in R Markdown. You can write code in dozens of languages and all do all kinds of clever tricks. You then run the code cell and any output the code generates, such as text or figures, will be displayed beneath the cell. We will get back to this in much more detail, but for now it's enough to understand that code cells are for executing code that is interpreted by a kernel (in this case the Python version in your Conda environment).
Before we continue, here are some shortcuts that can be useful. Note that they are only applicable when in command mode (blue frames). Most of them are also available from the menus. These shortcuts are also available from the Help menu in your notebook (there's even an option there to edit shortcuts).
| Shortcut | Effect |
|---|---|
| Enter | enter Edit mode |
| Esc | enter Command mode |
| Ctrl+Enter | run the cell |
| Shift+Enter | run the cell and select the cell below |
| Alt+Enter | run the cell and insert a new cell below |
| Ctrl+S | save the notebook |
| Tab | for code completion or indentation |
| M/Y | toggle between Markdown and Code cells |
| D-D | delete a cell |
| A/B | insert cells above/below current cell |
| X/C/V | cut/copy/paste cells |
| O | toggle output of current cell |
Writing markdown
Let's use our first cell to create a header. Change the format from Code to Markdown using the drop-down list in the Notebook Toolbar, or by pressing the M key when in command mode. Double click on the cell, or hit Enter to enter editing mode (green frame) and input "# My notebook" ("#" is used in Markdown for header 1). Run the cell with Ctrl-Enter.
Tada!
Markdown is a simple way to structure your notebook into sections with descriptive notes, lists, links, images etc.
Below are some examples of what you can do in markdown. Paste all or parts of it into one or more cells in your notebook to see how it renders. Make sure you set the cell type to Markdown.
## Introduction
In this notebook I will try out some of the **fantastic** concepts of Jupyter
Notebooks.
## Markdown basics
Examples of text attributes are:
* *italics*
* **bold**
* `monospace`
Sections can be separated by horizontal lines.
---
Blockquotes can be added, for instance to insert a Monty Python quote:
Spam!
Spam!
Spam!
Spam!
See [here](https://jupyter-notebook.readthedocs.io/en/stable/examples/Notebook/Working%20With%20Markdown%20Cells.html) for more information.
Writing code
Now let's write some code! Since we chose a Python kernel, Python would be the native language to run in a cell. Enter this code in the second cell and run it:
print("Hello world!")
Note how the output is displayed below the cell. This interactive way of working is one of the things that sets Jupyter Notebook apart from RStudio and R Markdown. R Markdown is typically rendered top-to-bottom in one run, while you work in a Jupyter notebook in a different way. This has partly changed with newer versions of RStudio, but it's probably still how most people use the two tools.
What is a Jupyter notebook? Let's look a little at the notebook we're
currently working in. Jupyter Notebooks are autosaved every minute or so, so you
will already have it available. We can be a little meta and do this from within
the notebook itself. We do it by running some shell commands in the third code
cell instead of Python code. This very handy functionality is possible by
prepending the command with !. Try !ls to list the files in the current
directory.
Aha, we have a new file called Untitled.ipynb! This is our notebook. Look at
the first ten lines of the file by using !head Untitled.ipynb. Seems like it's
just a plain old JSON file. Since it's a text file it's suitable for version
control with for example Git. It turns out that Github and Jupyter notebooks are
the best of friends, as we will see more of later. This switching between
languages and whatever-works mentality is very prominent within the Jupyter
notebook community.
Variables defined in cells become variables in the global namespace. You can therefore share information between cells. Try to define a function or variable in one cell and use it in the next. For example:
def print_me(str):
print(str)
and
print_me("Hi!")
Your notebook should now look something like this.
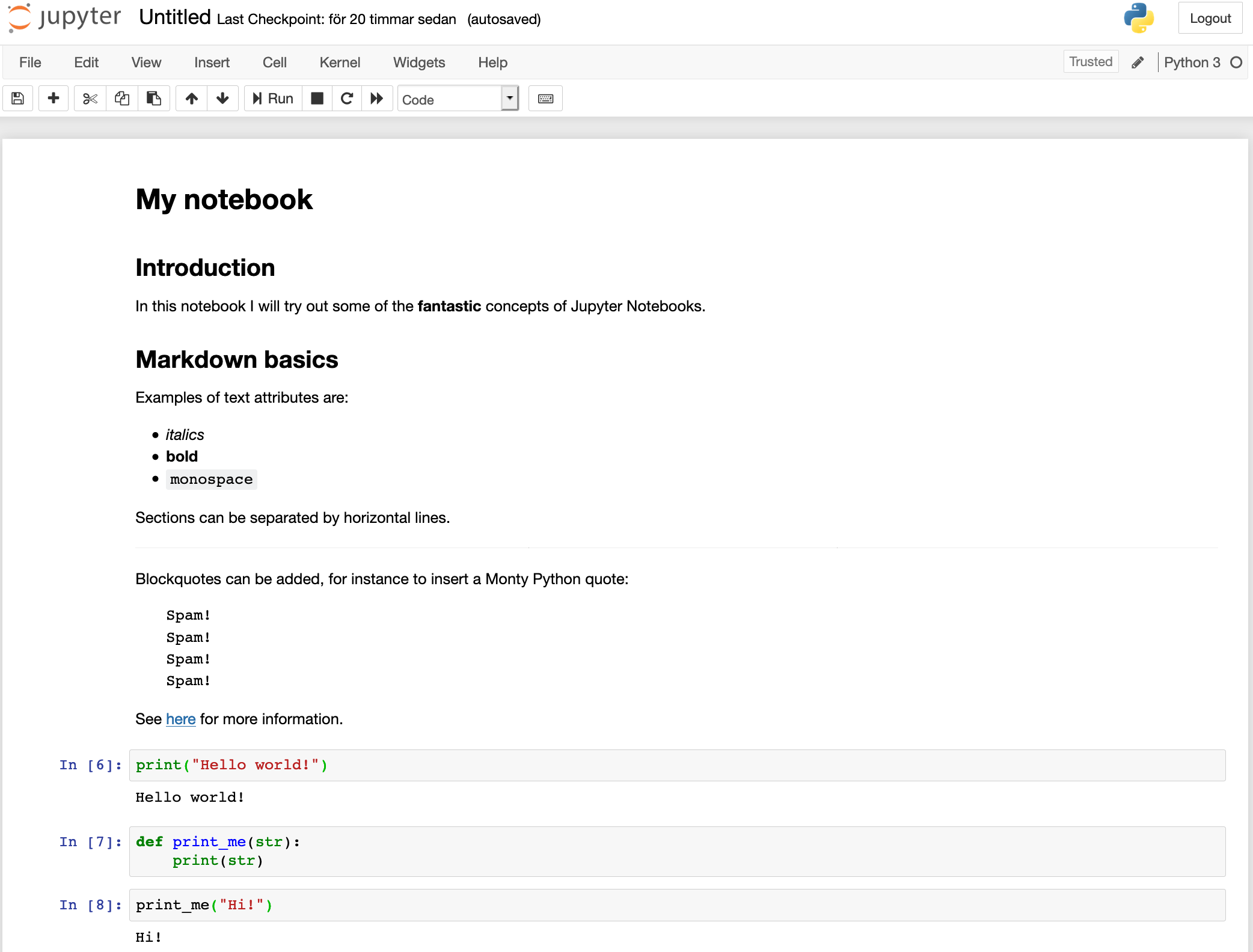
The focus here is not on how to write Markdown or Python; you can make really pretty notebooks with Markdown and you can code whatever you want with Python. Rather, we will focus on the Jupyter Notebook features that allow you to do a little more than that.
Quick recap
In this section we've learned:
- That a Jupyter notebook consists of a series of cells, and that they can be either markdown or code cells.
- That we execute the code in a code cell with the kernel that we chose when opening the notebook.
- We can run shell commands by prepending them with
!. - A Jupyter notebook is simply a text file in JSON format.
Magics
Magics constitute a simple command language that significantly extends the power of Jupyter notebooks. There are two types of magics:
- Line magics: Commands that are prepended by "%", and whose arguments only extend to the end of the line.
- Cell magics: Commands that start with
%%and then applies to the whole cell. Must be written on the first line of a cell.
Now list all available magics with %lsmagic (which itself is a magic). You
add a question mark to a magic to show the help (e.g. %lsmagic?). Some of
them act as shortcuts for commonly used shell commands (%ls, %cp, %cat,
..). Others are useful for debugging and optimizing your code (%timeit,
%debug, %prun, ..). For more information see the
magics documentation.
A very useful magic, in particular when using shell commands a lot in your
work, is %%capture. This will capture the stdout/stderr of any code cell and
store them in a Python object. Run %%capture? to display the help and try to
understand how it works. Try it out with either some Python code, other magics
or shell commands.
Click to see an example
%%capture output
%%bash
echo "Print to stdout"
echo "Print to stderr" >&2
and in another cell
print("stdout:" + output.stdout)
print("stderr:" + output.stderr)
Tip
You can capture the output of some magics directly like this:
my_dir = %pwd
print(my_dir)
The %%script magic is used for specifying a program (bash, perl, ruby, ..)
with which to run the code (similar to a shebang). For some languages it's
possible to use these shortcuts:
%%ruby%%perl%%bash%%html%%latex%%R(here you have to first install the rpy2 extension, for example with Conda, and then load with%load_ext rpy2.ipython)
Try this out if you know any of the languages above. Otherwise you can always try to print the quadratic formula with LaTeX!
\begin{array}{*{20}c} {x = \frac{{ - b \pm \sqrt {b^2 - 4ac} }}{{2a}}} & {{\rm{when}}} & {ax^2 + bx + c = 0} \\ \end{array}
Another useful magic is %precision which sets the floating point precision
in the notebook. As a quick example, add the following to a cell and run it:
float(100/3)
Next set the precision to 4 decimal points by running a cell with:
%precision 4
Now run the cell with float(100/3) again to see the difference.
Running %precision without additional arguments will restore the default.
Plotting
An essential feature of Jupyter Notebooks is of course the ability to visualize data and results via plots. A full guide to plotting in Python is beyond the scope of this course, but we'll offer a few glimpses into the plotting landscape of Python.
First of all, Python has a library for plotting called matplotlib, which comes packed with functionality for creating high-quality plots. Below is an example of how to generate a line plot of a sine wave.
# Import packages
import numpy as np
import matplotlib.pyplot as plt
# Generate a set of evenly spaced numbers between 0 and 100
x = np.linspace(0,3*np.pi,100)
# Use the sine function to generate y-values
y = np.sin(x)
# Plot the data
line, = plt.plot(x, y, color='red', linestyle="-")
By default plots are rendered in the notebook as rasterized images which can
make the quality poor. To render in scalable vector graphics format use the
set_matplotlib_formats function from the IPython package:
from IPython.display import set_matplotlib_formats
set_matplotlib_formats('pdf', 'svg')
Now try running the code for the sine wave plot again.
Other packages for plotting
As we mentioned Matplotlib comes with a lot of functionality which is great because it allows you to create all sorts of plots and modify them exactly to your liking. However, this can also mean that creating very basic plots might involve a lot of cumbersome coding, when all you want is a simple bar chart!
Fortunately there are a number of Python packages that build upon matplotlib but with a much simplified interface. One such popular package is seaborn. Below we'll see how to generate a nice looking bar plot with error bars.
First import the seaborn package (using an abbreviated name to simplify typing):
import seaborn as sns
Next we'll load some example data of penguins collected at the Palmer Station, in Antarctica.
penguins = sns.load_dataset("penguins")
# Look at first 5 lines of the data
penguins.head(5)
The most basic way to generate a bar plot of this data with seaborn is:
sns.barplot(data=penguins)
Simple right? Yes, but maybe not very informative. Here seaborn simply calculates the mean of all numeric variables for the penguins and plots them with error bars representing a 95% confidence interval.
Let's say that instead we want to plot the mean value of the body mass of the penguins at the different islands where they were examined.
sns.barplot(data=penguins, x="island", y="body_mass_g", ci="sd", errwidth=.5);
Here we specified to use values in the 'island' column as categories for the
x-axis, and values in the 'body_mass_g' column as values for the y-axis.
The barplot function of seaborn will then calculate the mean body mass for each
island and plot the bars. With ci="sd" we tell the function to draw the
standard deviation as error bars, instead of computing a confidence interval.
Finally errwidth=.5 sets the linewidth of the error bars.
If we instead want to visualize the data as a scatterplot we can use the
sns.scatterplot function. Let's plot the body mass vs. bill length for all
penguins and color the data points by species. We'll also move the legend
outside of the plotting area and modify the x and y-axis labels:
# Store the matplotlib axes containing the plot in a variable called 'ax'
ax = sns.scatterplot(data=penguins, x="bill_length_mm", y="body_mass_g",
hue="species")
# Modify the labels of the plot
ax.set_xlabel("Bill length (mm)")
ax.set_ylabel("Body mass (g)")
# Set legend position outside of plot
ax.legend(bbox_to_anchor=(1,1));
If you want to save a plot to file you can use the plt.savefig function. Add
the following to the bottom of the cell with the scatterplot code:
plt.savefig("scatterplot.pdf", bbox_inches="tight")
The bbox_inches="tight" setting ensures that the figure is not clipped when
saved to file.
The Seaborn website contains great tutorials and examples of other ways to plot data!
Interactive widgets
Since we're typically running our notebooks in a web browser, they are quite well suited for also including more interactive elements. A typical use case could be that you want to communicate some results to a collaborator or to a wider audience, and that you would like them to be able to modify how the results are displayed. It could, for example, be to select which gene to plot for, or to see how some parameter value affects a clustering. Jupyter notebooks has great support for this in the form of widgets.
Widgets are eventful Python objects that have a representation in the browser,
often as a control like a slider, textbox, etc. These are implemented in the
ipywidgets package.
The easiest way to get started with using widgets are via the interact and
interactive functions. These functions autogenerate widgets from functions
that you define, and then call those functions when you manipulate the widgets.
Too abstract? Let's put it into practice!
Let's try to add sliders that allow us to change the frequency, amplitude and phase of the sine curve we plotted previously.
# Import the interactive function from ipywidgets
from ipywidgets import interactive
# Also import numpy (for calculating the sine curve)
# and pyplot from matplotlib for plotting
import numpy as np
import matplotlib.pyplot as plt
# Define the function for plotting the sine curve
def sine_curve(A, f, p):
# Set up the plot
plt.figure(1, figsize=(4,4))
# Create a range of 100 evenly spaced numbers between 0 and 100
x = np.linspace(0,10,100)
# Calculate the y values using the supplied parameters
y = A*np.sin(x*f+p)
# Plot the x and y values ('r-' specifies color and line style)
plt.plot(x, y, color='red', linestyle="-")
# Here we supply the sine_curve function to interactive,
# and set some limits on the input parameters
interactive_plot = interactive(sine_curve,
A=(1, 5, 1),
f=(0, 5, 1),
p=(1, 5, 0.5))
# Display the widgets and the plot
interactive_plot
The code above defines a function called sine_curve which takes three
arguments:
A= the amplitude of the curvef= the frequency of the curvep= the phase of the curve
The function creates a plot area, generates x-values and calculates y-values
using the np.sin function and the supplied parameters. Finally, the x and y
values are plotted.
Below the function definition we use interactive with the sine_curve
function as the first parameter. This means that the widgets will be tied to
the sine_curve function. As you can see we also supply the A, f and p
keyword arguments. Importantly, all parameters defined in the sine_curve
function must be given in the interactive call and a widget is created for
each one.
Depending on the type of the passed argument different types of
widgets will be created by interactive. For instance:
intorfloatarguments will generate a sliderboolarguments (True/False) will generate checkbox widgetslistarguments will generate a dropdownstrarguments will generate a text-box
By supplying the arguments in the form of
tuples we can
adjust the properties of the sliders. f=(1, 5, 1) creates a widget with
minimum value of 1, maximum value of 5 and a step size of 1. Try adjusting
these numbers in the interactive call to see how the sliders change (you have
to re-execute the cell).
The final line of the cell (interactive_plot) is where the actual widgets and
plot are displayed. This code can be put in a separate cell, so that you can
define functions and widgets in one part of your notebook, and reuse them
somewhere else.
This is how it should look if everything works. You can now set the frequency amplitude and phase of the sine curve by moving the sliders.
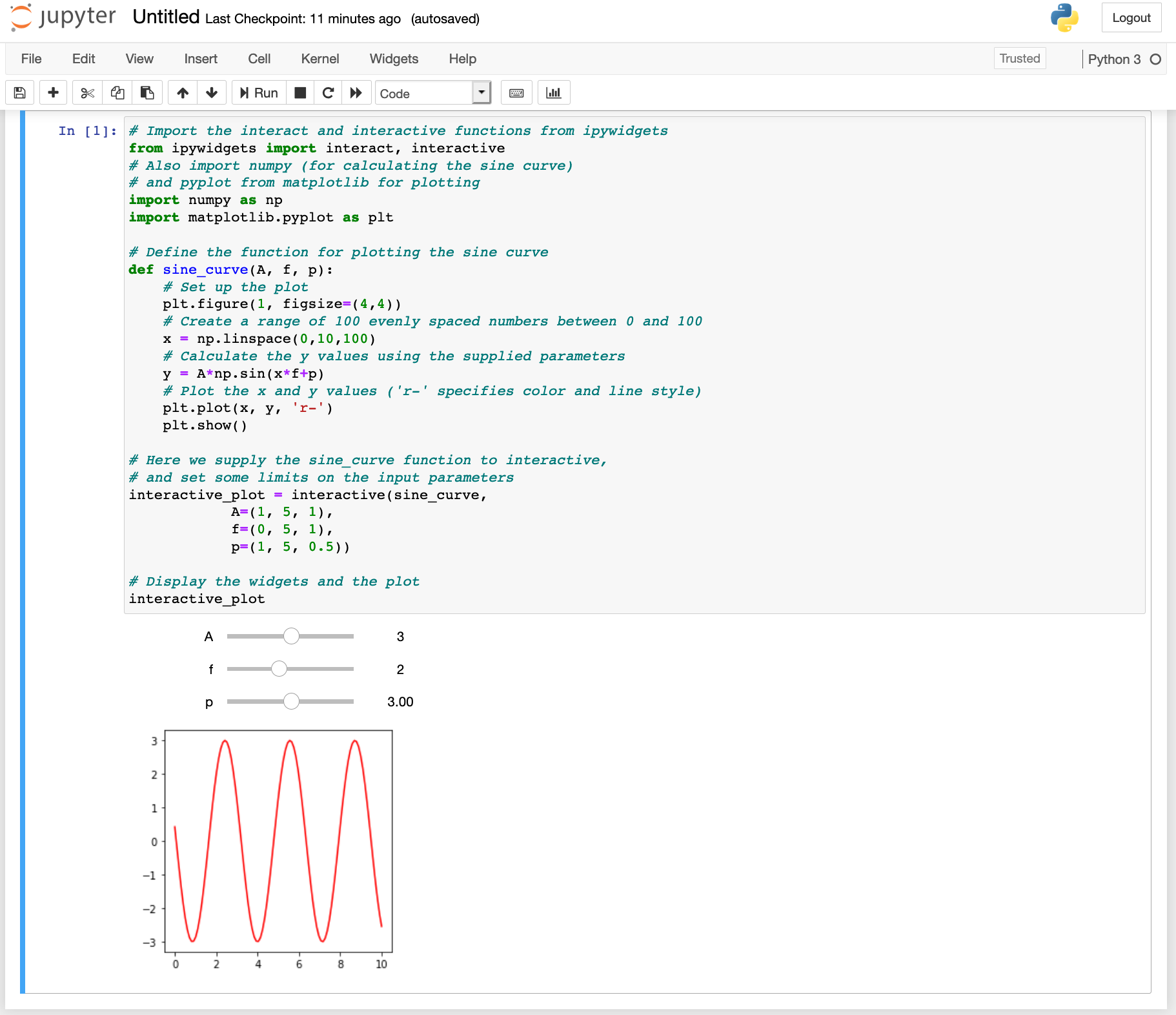
There are lots of widgets, e.g.:
- Dropdown menus
- Toggle buttons
- Range sliders
- File uploader
... and much, much more. Here is a list of all available widgets
together with documentation and examples. Some of these widgets cannot be
autogenerated by interactive, but fear not! Instead of relying on
autogeneration we can define the widget and supply it directly to interactive.
To see this in practice, change out the A argument to a pre-defined
IntSlider widget. First define the slider:
from ipywidgets import widgets
A = widgets.IntSlider(value=2, min=1, max=5, step=1)
Then replace the call to interactive so that it looks like this:
interactive_plot = interactive(sine_curve, A=A, f=5, p=5)
Extra challenge
If you can't get enough of widgets you might want to try this out: see if you
can figure out how to add a widget that lets you pick the color for the sine
curve line. Search for the appropriate widget in the Widget list.
You'll need to update the sine_curve function and pass the new widget as
an argument in the call to interactive. If you need help, click below.
Click to see how to add a color picker
# Import the interactive function from ipywidgets
from ipywidgets import interactive
# Also import numpy (for calculating the sine curve)
# and pyplot from matplotlib for plotting
import numpy as np
from ipywidgets import widgets ## <- import widgets
import matplotlib.pyplot as plt
# Define the function for plotting the sine curve
def sine_curve(A, f, p, color): ## <- add parameter here
# Set up the plot
plt.figure(1, figsize=(4,4))
# Create a range of 100 evenly spaced numbers between 0 and 100
x = np.linspace(0,10,100)
# Calculate the y values using the supplied parameters
y = A*np.sin(x*f+p)
# Plot the x and y values
plt.plot(x, y, color=color) ## <- Use color from widget here
# Here we supply the sine_curve function to interactive,
# and set some limits on the input parameters
# Define the colorpicker widget
colorpicker = widgets.ColorPicker(description='color',value="red")
interactive_plot = interactive(sine_curve,
A=(1, 5, 1),
f=(0, 5, 1),
p=(1, 5, 0.5),
color=colorpicker) ## <- Supply the colorpicker to the function
# Display the widgets and the plot
interactive_plot
Color picking
Note that you may have to close the color picker once you've made your choice in order to make the plot update.
Other interactive plots
Jupyter widgets, like we used here, is the most vanilla way of getting interactive graphs in Jupyter notebooks. Some other alternatives are:
- Plotly is actually an API to a web service that renders your graph and returns it for display in your Jupyter notebook. Generates very visually appealing graphs, but from a reproducibility perspective it's maybe not a good idea to be so reliant on a third party.
- Bokeh is another popular tool for interactive graphs. Most plotting packages for Python are built on top of matplotlib, but Bokeh has its own library. This can give a steeper learning curve if you're used to the standard packages.
- mpld3 tries to integrate matplotlib with Javascript and the D3js package. It doesn't scale well for very large datasets, but it's easy to use and works quite seamlessly.
Quick recap
In the three previous sections we've learned:
- How magics can be used to extend the power of Jupyter notebooks, and the difference between line magics and cell magics.
- How to switch between different languages by using magics.
- How to do some basic plotting in Jupyter.
- How to implement interactive widgets.
Jupyter extensions
Jupyter Notebook extensions are add-ons that can increase the functionality of
your notebooks. These were installed in the Setup section of this
tutorial by including the jupyter_contrib_nbextensions package in the conda
environment file. You can read more about the extensions
here.
To manage extensions go to the Jupyter dashboard in your browser and click the Nbextensions tab. You should see something similar to this:
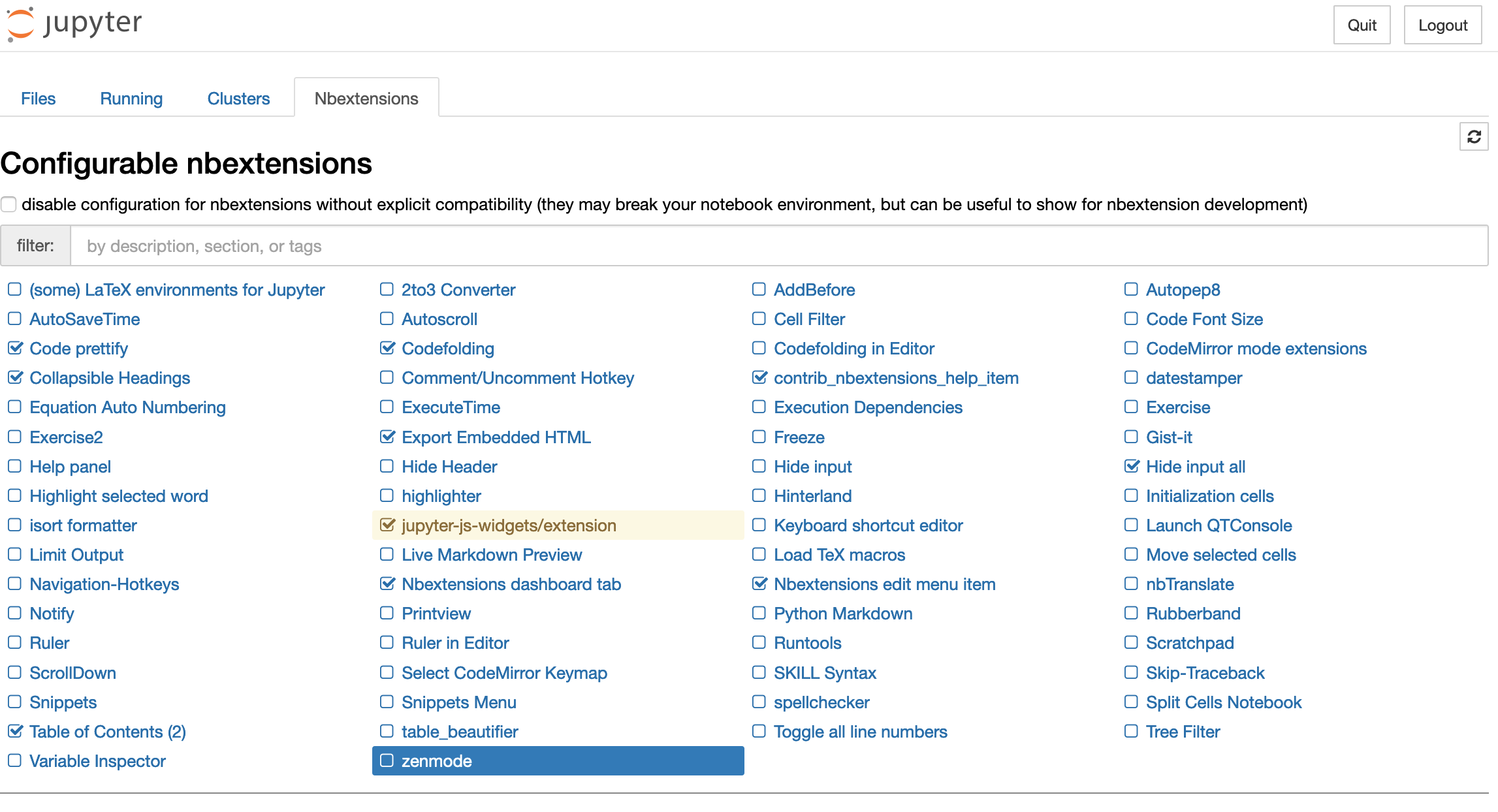
Clicking an extension in the list displays more information about it. To enable/disable extensions simply click the checkbox next to the extension name in the list. Some useful extensions include
-
Hide input all, which allows you to hide all code cells with the click of a button.
-
Collapsible Headings, which allows you to collapse sections below markdown headings to increase readability.
-
Table of Contents (2), which adds a table of contents to the notebook making navigation a lot quicker especially for long notebooks.
Feel free to peruse the list and find your own favourites! Keep in mind that these are unofficial, community-contributed extensions and as such they come with few, if any, guarantees.
Using the command line
Converting notebooks
Notebooks can be converted to various output formats such as HTML, PDF, LaTeX etc. directly from the File -> Download as menu.
Conversion can also be performed on the command line using the jupyter
nbconvert command. nbconvert is installed together with the jupyter Conda
package and is executed on the command line by running jupyter nbconvert.
The syntax for converting a Jupyter notebook is:
jupyter nbconvert --to <FORMAT> notebook.ipynb
Here <FORMAT> can be any of asciidoc, custom, html, latex, markdown,
notebook, pdf, python, rst, script, slides. Converting to some
output formats (e.g. PDF) may require you to install separate software such
as Pandoc or a TeX environment.
Try converting the Untitled.ipynb notebook that you have been working on so
far to HTML using jupyter nbconvert.
Tip
To export notebooks in the form they appear with Jupyter Extensions activated
you can make use of the nbextensions template that is installed with the
jupyter_contrib_nbextensions package. Adding --template=nbextensions to
the jupyter nbconvert call should do the trick, but note that not all
extensions are guaranteed to display right after exporting.
Executing notebooks
nbconvert can also be used to run a Jupyter notebook from the commandline. By
running:
jupyter nbconvert --execute --to <FORMAT> notebook.ipynb
nbconvert executes the cells in a notebook, captures the output and saves the
results in a new file. Try running it on the Untitled.ipynb notebook.
You can also specify a different output file with --output <filename>.
So in order to execute your Untitled.ipynb notebook and save it to a file
named report.html you could run:
jupyter nbconvert --to html --output report.html --execute Untitled.ipynb
Jupyter and the case study
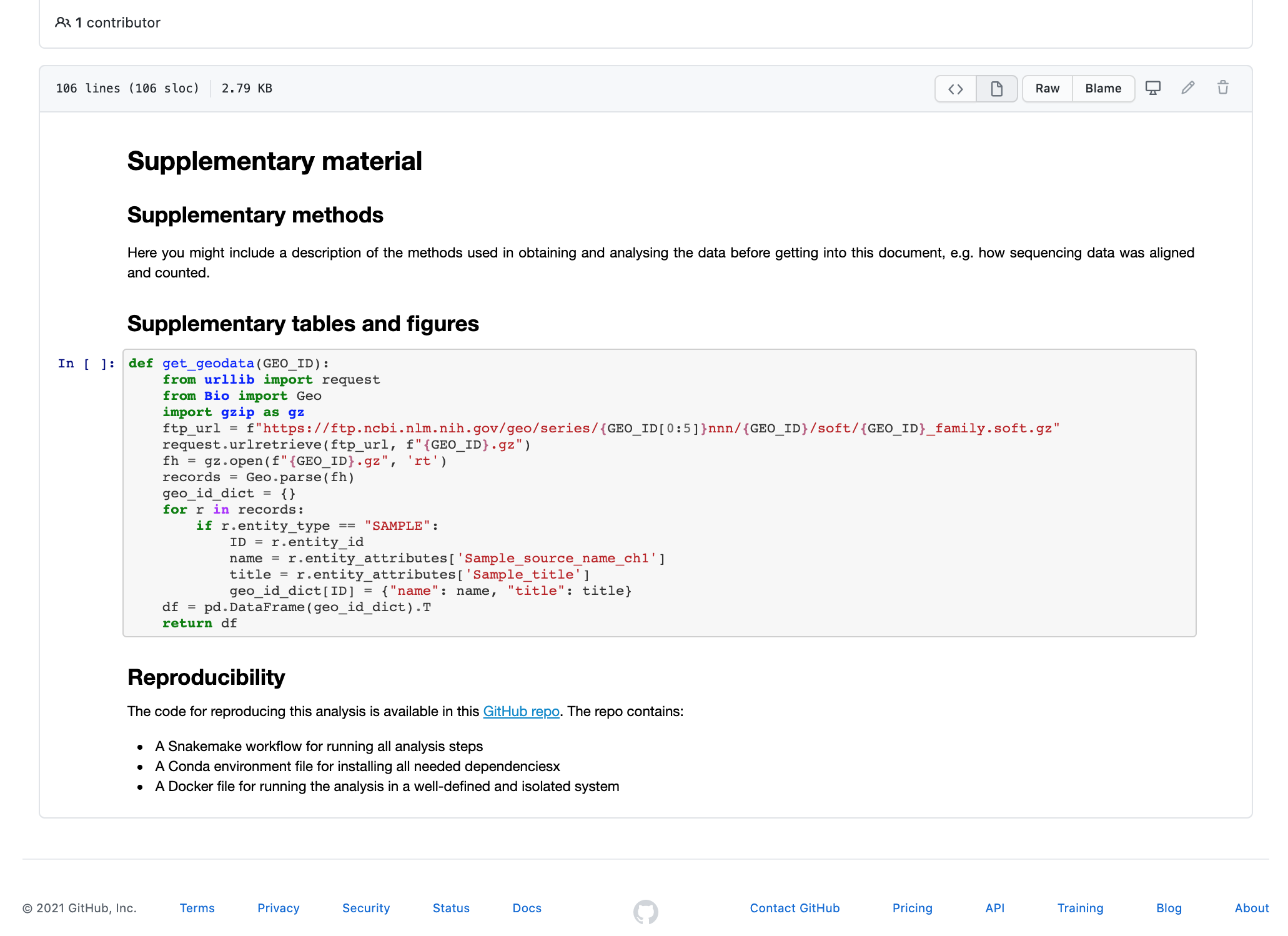
As you might remember from the intro, we are attempting to understand how lytic bacteriophages can be used as a future therapy for the multiresistant bacteria MRSA (methicillin-resistant Staphylococcus aureus). We have already seen how to define the project environment in the Conda tutorial and how to set up the workflow in the Snakemake tutorial. Here we explore the results from the Snakemake tutorial and generate a Supplementary Material file with some basic stats.
In the jupyter/ directory you will find a notebook called
supplementary_material.ipynb. Open this notebook with jupyter by running:
jupyter notebook supplementary_material.ipynb
Tip
Using what you've learned about markdown in notebooks, add headers and descriptive text to subdivide sections as you add them. This will help you train how to structure and keep note of your work with a notebook.
You will see that the notebook contains only a little markdown text and a code
cell with a function get_geodata. We'll start by adding a cell with some
import statements.
Create a new cell after the get_geodata function but
before the Reproducibility section and add the following to it:
import pandas as pd
import seaborn as sns
import matplotlib.pyplot as plt
import numpy as np
pandas (for working with tables), seaborn and
matplotlib.pyplot (for plotting) and numpy (for numerical operations)
Python modules.
Also add:
from IPython.display import set_matplotlib_formats
set_matplotlib_formats('pdf', 'svg')
to set high-quality output for plots.
Run the cell and create a new one below it.
In the next cell we'll define some parameters to use for the notebook:
counts_file="results/tables/counts.tsv"
multiqc_file="intermediate/multiqc_general_stats.txt"
rulegraph_file="results/rulegraph.png"
SRR_IDs=["SRR935090","SRR935091","SRR935092"]
GSM_IDs=["GSM1186459","GSM1186460","GSM1186461"]
GEO_ID="GSE48896"
As you can see we add paths to results files and define lists with some sample IDS. Run this cell and add a new one below it.
Next, we'll fetch some sample information from NCBI using the get_geodata
function defined at the start of the notebook and collate it into a dataframe.
id_df = pd.DataFrame(data=GSM_IDs, index=SRR_IDs, columns=["geo_accession"])
geo_df = get_geodata(GEO_ID)
name_df = pd.merge(id_df, geo_df, left_on="geo_accession", right_index=True)
# Create a dictionary to rename sample ids in downstream plots
name_dict = name_df.to_dict()
Take a look at the contents of the name_df dataframe (e.g. run a cell with
that variable only to output it below the cell).
Now we'll load some statistics from the QC part of the workflow, specifically
the 'general_stats' file from multiqc. Add the following to a new cell and run
it:
qc = pd.read_csv(multiqc_file, sep="\t")
qc.rename(columns=lambda x: x.replace("FastQC_mqc-generalstats-fastqc-", "").replace("_", " "), inplace=True)
qc = pd.merge(qc, name_df, left_on="Sample", right_index=True)
qc
In the code above we load the multiqc file, rename the columns by stripping the
FastQC_mqc-generalstats-fastqc- part from column names and replace underscores
with spaces. Finally the table is merged with the information obtained in the
step above and output to show summary statistics from the QC stage.
Next it's time to start loading gene count results from the workflow. Start by reading the counts file, and edit the columns and index:
# Read count data
counts = pd.read_csv(counts_file, sep="\t", header=0)
# Rename columns to extract SRR ids
counts.rename(columns = lambda x: x.split("/")[-1].replace(".sorted.bam",""), inplace=True)
# Set index to gene ids
gene_names = dict(zip([x[0] for x in counts.index], [x[1] for x in counts.index]))
counts.index = [x[0] for x in counts.index]
Take a look at the counts dataframe to get an idea of the data structure. As
you can see the dataframe shows read counts for genes (rows) in each sample
(columns).
The last few rows that are prefixed with '__' correspond to summary
statistics output from htseq-count for unassigned reads. We'll extract
these lines from the dataframe for downstream visualization:
# Extract stats from htseq starting with "__"
counts_other = counts.loc[counts.index.str.startswith("__")]
counts_other = counts_other.rename(index=lambda x: x.lstrip("_"))
# Drop the "__" lines from counts
counts = counts.drop(counts.loc[counts.index.str.startswith("__")].index)
Now let's generate a barplot showing number of reads assigned to genes as well as reads unassigned for various reasons. First we sum up all assigned reads per sample and merge it with the unassigned stats from the previous step:
# Sum counts in 'genes' and merge with 'other' categories
count_data = pd.DataFrame(counts.sum(), columns = ["genes"])
count_data = pd.merge(count_data, counts_other.T, left_index=True, right_index=True)
Now for the plotting:
# Set color palette to 'husl', with number of colors corresponding to categories
# in the count_data
colors = sns.color_palette("husl", n_colors=count_data.shape[1])
# Create a stacked barplot
ax = count_data.plot(kind="bar", stacked=True, color=colors)
# Move legend and set legend title
ax.legend(bbox_to_anchor=(1,1), title="Feature");
The final plot will be a heatmap of gene counts for a subset of the genes. We'll select genes whose standard deviation/mean count across samples is greater than 1.2, and have a maximum of at least 5 reads in 1 or more sample:
heatmap_data = counts.loc[(counts.std(axis=1).div(counts.mean(axis=1))>1.2)&(counts.max(axis=1)>5)]
In order to make the heatmap more informative we'll also add gene names to the rows of the heatmap data, and replace the SRR ids with the title of samples used in the study:
heatmap_data = heatmap_data.rename(index=lambda x: f"{x} ({gene_names[x]})")
heatmap_data.rename(columns = lambda x: name_dict['title'][x], inplace=True)
Now let's plot the heatmap. We'll log-transform the counts, set color scale to Blue-Yellow-Red and cluster both samples and genes using 'complete' linkage clustering:
with sns.plotting_context("notebook", font_scale=0.7):
ax = sns.clustermap(data=np.log10(heatmap_data+1), cmap="RdYlBu_r",
method="complete", yticklabels=True, linewidth=.5,
cbar_pos=(0.2, .8, 0.02, 0.15), figsize=(8,6))
plt.setp(ax.ax_heatmap.get_xticklabels(), rotation=270)
In the code above we use the seaborn plotting_context function to scale all
text elements of the heatmap in one go.
As a final step we'll add some info for reproducibility under the
Reproducibility section. To add the overview image of the workflow found in
results/rulegraph.png we can use the Image function from IPython.display:
from IPython.display import Image
Image(rulegraph_file)
Let's also output the full conda environment so that all packages and versions are included in the notebook. There are several ways this can be done, for example you could simply add:
!conda list
to the end of the notebook.
Snakemake integration
If you want to know more about how notebooks can be integrated into Snakemake worfklows, see the Extra material at the end of this tutorial
Sharing your work
The files you're working with come from a GitHub repo. Both GitHub and Bitbucket
can render Jupyter notebooks as well as other types of Markdown documents. Now
go to our GitHub repo at
https://github.com/NBISweden/workshop-reproducible-research
and navigate to jupyter/supplementary_material.ipynb.
As you can imagine, having this very effortless way of sharing results can greatly increase the visibility of your work. You work as normal on your project, and push regularly to the repository as you would anyways, and the output is automatically available for anyone to see. Or for a select few if you're not ready to share your findings with the world quite yet.
Say your notebook isn't on Github/Bitbucket. All hope isn't lost there. Jupyter.org provides a neat functionality called nbviewer, where you can past an URL to any notebook and they will render it for you. Go to https://nbviewer.jupyter.org and try this out with our notebook.
https://raw.githubusercontent.com/NBISweden/workshop-reproducible-research/main/jupyter/supplementary_material.ipynb
Shared interactive notebooks
So far we've only shared static representations of notebooks. A strong trend at the moment is to run your notebooks in the cloud, so that the person you want to share with could actually execute and modify your code. This is a great way of increasing visibility and letting collaborators or readers get more hands-on with your data and analyses. From a reproducibility perspective, there are both advantages and drawbacks. On the plus side is that running your work remotely forces you to be strict when it comes to defining the environment it uses (probably in the form of a Conda environment or Docker image). On the negative side is that you become reliant on a third-party service that might change input formats, go out of business, or change payment model.
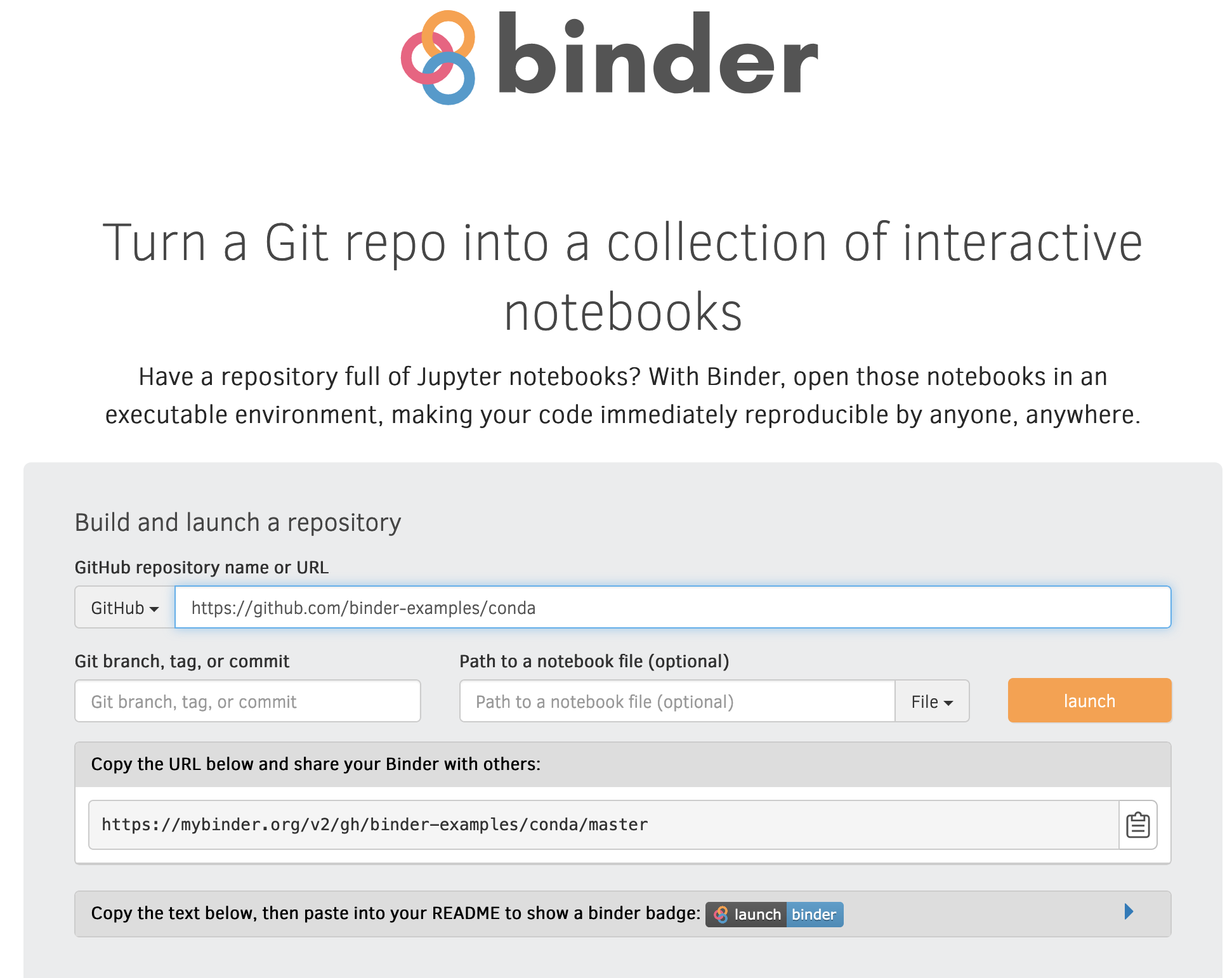
Here we will try out a service called Binder, which lets you run and
share Jupyter Notebooks in Git repositories for free. There are a number
of example repositories that are
setup to be used with Binder. Navigate to
https://github.com/binder-examples/conda/
to see one such example. As you can see the repository contains a LICENSE
file, a README, an environment file and a notebook. To use a repository
with Binder the environment file should contain all the packages needed
to run notebooks in the repo. So let's try to run the index.ipynb file
using Binder:
Just go to https://mybinder.org and paste the link to the GitHub repo. Note the link that you can use to share your notebook. Then press "launch".
What will happen now it that:
- Binder detects the
environment.ymlfile in the root of the repo. Binder then builds a Docker image based on the file. This might take a minute or two. You can follow the progress in the build log. - Binder then launches the Jupyter Notebook server in the Docker container..
- ..and opens a browser tab with it for you.
Once the process is finished you will be presented with a Jupyter server
overview of the contents in the repository. Click on the index.ipynb
notebook to open it. Tada! You are now able to interact with (and
modify) someone else's notebook online.
Applied to your own projects you now have a way to run analyses in the cloud and in an environment that you define yourself. All that's needed for someone to replicate your analyses is that you share a link with them. Note that notebooks on Binder are read-only; its purpose is for trying out and showing existing notebooks rather than making new ones.
Binder configuration files
By default Binder looks for configuration files such as environment.yml
in the root of the repository being built. But you may also put
such files outside the root by making a binder/ folder in the root
and placing the file there.
A note on transparency
Resources like Github/Bitbucket and Jupyter Notebooks have changed the way we do scientific research by encouraging visibility, social interaction and transparency. It was not long ago that the analysis scripts and workflows in a lab were well-guarded secrets that we only most reluctantly shared with others. Assuming that it was even possible. In most cases, the only postdoc who knew how to get it to work had left for a new position in industry, or no one could remember the password to the file server. If you're a PhD student, we encourage you to embrace this new development wholeheartedly, for it will make your research better and make you into a better scientist. And you will have more fun.
Extra material
Running jupyter notebooks on a cluster
-
Login to Uppmax, making sure to use a specific login node, e.g.
rackham1:ssh <your-user-name>@rackham1.uppmax.uu.se -
Create/activate a conda environment containing
jupyterthen runpythonto start a Python console. Type:
import IPython.lib
IPython.lib.passwd()
'sha1:'
* Create a config file named e.g. my_jupyter_config.py and add this to it:
c = get_config()
# Notebook config
#c.NotebookApp.certfile = u''
c.NotebookApp.ip = 'localhost'
c.NotebookApp.open_browser = False
c.NotebookApp.password = u'sha1:...' #<-- Update with your 'sha1:' string here
c.NotebookApp.port = 9990
jupyter notebook --config my_jupyter_config.py
On your local computer * Forward port 8080 to the remote port on the Uppmax login node:
ssh -N -f -L localhost:8080:localhost:9990 <your-user-name>@rackham1.uppmax.uu.se
- Connect to the jupyter server by opening
localhost:8080in your browser. You should be prompted for the password you generated.
You are now (hopefully) accessing the jupyter server that's running on Upmmax, via your local browser.
Integrating notebooks with Snakemake workflows
In the case study section of this tutorial we
created a Jupyter notebook that used output from a Snakemake workflow
and produced some summary results and plots. Wouldn't it be nice if this was
actually part of the workflow itself? To generate a HTML version of the notebook
we can use what we learned in the section about
Converting noteboks. The command to execute the notebook
and save it in HTML format in a file results/supplementary.html would be:
jupyter nbconvert --to HTML --output-dir results --output supplementary.html --execute supplementary_material.ipynb
This command could be used in a rule, e.g. make_supplementary, the input of
which would be results/tables/counts.tsv, intermediate/multiqc_general_stats.txt,
and results/rulegraph.png. See if you can work out how to implement such a
rule at the end of the Snakefile found in the jupyter/ directory. Click
below to see an example.
Click to see an example of the make_supplementary rule
rule make_supplementary:
input:
counts = "results/tables/counts.tsv",
multiqc_file = "intermediate/multiqc_general_stats.txt",
rulegraph = "results/rulegraph.png"
output:
"results/supplementary.html"
params:
base = lambda wildcards, output: os.path.basename(output[0]),
dir = lambda wildcards, output: os.path.dirname(output[0])
shell:
"""
jupyter nbconvert --to HTML --output-dir {params.dir} --output {params.base} \
--execute supplementary_material.ipynb
"""
Note
The conda enivronment for the jupyter tutorial does not contain packages
required to run the full snakemake workflow. So if you wish to test jupyter
integration fully you should update the conda environment by running
conda install snakemake-minimal fastqc sra-tools multiqc bowtie2 tbb
samtools htseq bedtools wget graphviz
Moar integration!
Snakemake actually supports the execution of notebooks via the notebook:
rules directive. See more about Jupyter integration in the
snakemake docs.
In the notebook: directive of such a rule you specify the path to a jupyter
notebook (relative to the Snakefile) which is then executed when
the rule is run.
So how is this useful?
In the notebook itself this gives you access to a snakemake object
containing information about input and output files for the rule via
snakemake.input and snakemake.output. Similarly, you can access rule
wildcards with snakemake.wildcards, params with snakemake.params,
and config settings with snakemake.config.
When snakemake runs the rule with the notebook: directive jupyter-nbconvert
is used to execute the notebook. No HTML output is generated here but it is
possible to store a version of the notebook in its final processed form by
adding
log:
notebook="<path>/<to>/<processed>/<notebook.ipynb>"
to the rule.
Because you won't get the notebook in full HTML glory, this type of integration is better suited if you want to use a notebook to generate figures and store these in local files (e.g. pdf/svg/png formats).
We'll use the supplementary_material.ipynb notebook as an example! Let's say
that instead of exporting the entire notebook to HTML we want a rule that
outputs pdf versions of the barplot and heatmap figures we created.
Let's start by setting up the rule. For simplicity we'll use the same input as
when we edited the notebook in the first place. The output will be
results/barplot.pdf and results/heatmap.pdf. Let's also output a finalized
version of the notebook using the log: notebook= directive:
rule make_supplementary_plots:
input:
counts = "results/tables/counts.tsv",
multiqc = "intermediate/multiqc_general_stats.txt",
rulegraph = "results/rulegraph.png"
output:
barplot = "results/barplot.pdf",
heatmap = "results/heatmap.pdf"
log:
notebook = "results/supplementary.ipynb"
The notebook will now have access to snakemake.input.counts,
snakemake.output.barplot and snakemake.output.heatmap when executed from
within the workflow. Let's go ahead and edit the notebook! In the cell where we
defined notebook parameters edit the code so that it looks like this:
counts_file=snakemake.input.counts
multiqc_file=snakemake.input.multiqc
rulegraph_file=snakemake.input.rulegraph
SRR_IDs=snakemake.params.SRR_IDs
GSM_IDs=snakemake.params.GSM_IDs
GEO_ID=snakemake.params.GEO_ID
Notice that we set the SRR_IDs, GSM_IDs and GEO_ID variables using
variables in snakemake.params? However, we haven't defined these in our rule
yet so let's go ahead and do that now. Add the params section so that the
make_supplementary_plots in the Snakefile looks like this:
rule make_supplementary_plots:
input:
counts = "results/tables/counts.tsv",
multiqc = "intermediate/multiqc_general_stats.txt",
rulegraph = "results/rulegraph.png"
output:
barplot = "results/barplot.pdf",
heatmap = "results/heatmap.pdf"
log:
notebook = "results/supplementary.ipynb"
params:
SRR_IDs = ["SRR935090","SRR935091","SRR935092"],
GSM_IDs = ["GSM1186459", "GSM1186460", "GSM1186461"],
GEO_ID = "GSE48896"
notebook: "supplementary_material.ipynb"
Tip
One way to further generalize this rule could be to define the SRR_IDs, GSM_IDs
and GEO_ID parameters in a config file instead, in which case they would be
directly accessible from within the notebook using snakemake.config['SRR_IDs']
etc.
Now the rule contains everything needed, but we still need to edit the notebook to save the plots to the output files. First, edit the cell that generates the barplot so that it looks like this:
count_data = pd.DataFrame(counts.sum(), columns = ["genes"])
count_data = pd.merge(count_data, counts_other.T, left_index=True, right_index=True)
colors = sns.color_palette("husl", n_colors=count_data.shape[1])
ax = count_data.plot(kind="bar", stacked=True, color=colors)
ax.legend(bbox_to_anchor=(1,1), title="Feature");
plt.savefig(snakemake.output.barplot, dpi=300, bbox_inches="tight") ## <-- Add this line!
Finally, edit the cell that generates the heatmap so that it looks like this:
heatmap_data = counts.loc[(counts.std(axis=1).div(counts.mean(axis=1))>1.2)&(counts.max(axis=1)>5)]
heatmap_data = heatmap_data.rename(index=lambda x: f"{x} ({gene_names[x]})")
heatmap_data.rename(columns = lambda x: name_dict['title'][x], inplace=True)
with sns.plotting_context("notebook", font_scale=0.7):
ax = sns.clustermap(data=np.log10(heatmap_data+1), cmap="RdYlBu_r",
method="complete", yticklabels=True, linewidth=.5,
cbar_pos=(0.2, .8, 0.02, 0.15), figsize=(8,6))
plt.setp(ax.ax_heatmap.get_xticklabels(), rotation=270)
plt.savefig(snakemake.output.heatmap, dpi=300, bbox_inches="tight") ## <-- Add this line!
Now you can run the following to generate the plots:
snakemake -j 1 make_supplementary_plots
Presentations with Jupyter
As if all the above wasn't enough you can also create presentations/slideshows with Jupyter! Simply use conda to install the RISE extension to your jupyter environment:
conda install -c conda-forge rise
then open up a notebook of your choice. In the menu click View->Cell Toolbar->Slideshow. Now every cell will have a drop-down in the upper right corner allowing you to set the cell type:
- Slide: a regular slide
- Sub-Slide: a regular slide that will be displayed below the previous
- Fragment: these cells split up slides so that content (fragments) are added only when you press Space
- Skip: these cells will not appear in the presentation
- Notes: these cells act as notes, shown in the speaker view but not in the main view
The presentation can be run directly from the notebook by clicking the
'Enter/Exit RISE Slideshow' button (looks like a bar chart) in the toolbar, or
by using the keyboard shortcut Alt-r. Running it directly from a notebook
means you can also edit and run cells during your presentation. The downside is
that the presentation is not as portable because it may rely on certain software
packages that other people are not comfortable with installing.
You can also export the notebook to an HTML-file with jupyter nbconvert
--execute --to SLIDES <your-notebook.ipynb>. The resulting file, with the
slideshow functionality included, can be opened in any browser. However, in
this format you cannot run/edit cells.